- Genome assembly with 2nd and 3rd WGS data
- Software installation
- Exploration with a demo data
Genome assembly with 2nd and 3rd WGS data
In this session, we aim to assemble a bacterial genome using 2nd and 3rd whole genome sequencing (WGS) data. We will use it this as example to explore the WGS data analysis, and look into the difference between sequencing technologies.
Software installation
Docker image
You can pull the image from DockerHub with the following command:
docker pull yanhui09/mac2023_extra
Installation with mamba
It’s recommended to install the software with mamba in an independent conda environment.
Assuming you have installed mamba in your system, you can create a new conda environment with mamba and install the software with the following commands:
Go to the downloaded data directory. Make sure you know where you are in the terminal.
E.g., The downloaded data directory is placed at /home/username/MAC2023-extera.
cd /home/username/MAC2023-extera
pwd
You shall see the path of the downloaded data directory as below.
/home/username/MAC2023-extera
Now you can create a new conda environment with mamba and install the software with the following commands:
mamba env create -n wgs1 -f envs/env1.yaml
Activate the environment for the following analysis.
conda activate wgs1
Exploration with a demo data
The WGS data can be fetched from MAC2023-extra as before. You shall download the data already if you have completed the previous exercises. In case you haven’t, you can refer to the requisites section.
First look at the data with seqkit and fastqc
seqkit stat data/wgs/*.fastq.gz data/wgs/ncbi_pacbio_TL110.fasta
Expected output:
file format type num_seqs sum_len min_len avg_len max_len
data/wgs/NXT20x_R1.fastq.gz FASTQ DNA 200,000 30,200,000 151 151 151
data/wgs/NXT20x_R2.fastq.gz FASTQ DNA 200,000 30,200,000 151 151 151
data/wgs/ont_r10_20x.fastq.gz FASTQ DNA 7,862 51,201,670 129 6,512.6 87,688
data/wgs/ncbi_pacbio_TL110.fasta FASTA DNA 1 2,566,312 2,566,312 2,566,312 2,566,312
Here is a set of sequencing data from a Propionibacterium freudenreichii strain. We subsampled the sequencing data to 20x coverage for the Illumina and Oxford Nanopore Technologies(ONT) reads. The PacBio reference genome is from the NCBI RefSeq database.
Q1: What is the genome size of this strain? How is the sequencing coverage calculated?
illumina: 30,200,000*2/2,566,312≈20
ONT: 51,201,670/2,566,312≈20
Let’s have a look at the quality of the sequencing data with fastqc.
mkdir -p fastqc/illumina fastqc/ont_r10
fastqc data/wgs/NXT20x_R*.fastq.gz -o ./fastqc/illumina
fastqc data/wgs/ont_r10_20x.fastq.gz -o ./fastqc/ont_r10
You can open the .html files in the fastqc directory to have a look at the quality of the sequencing data.
Illumina 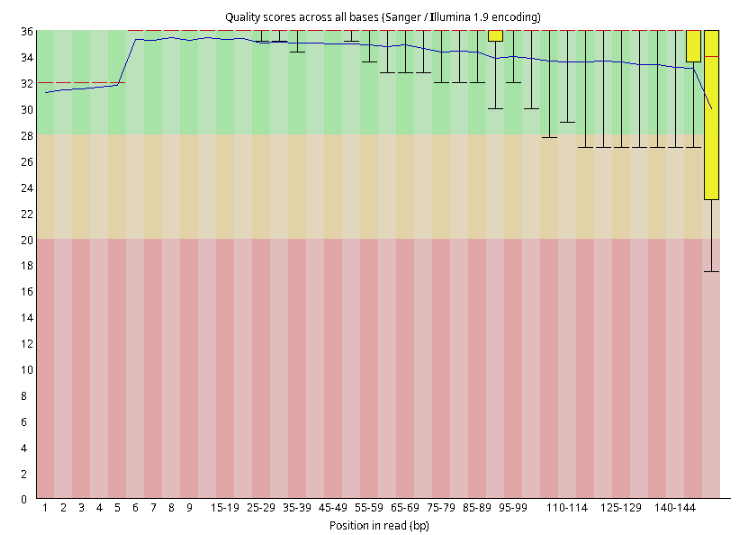
ONT 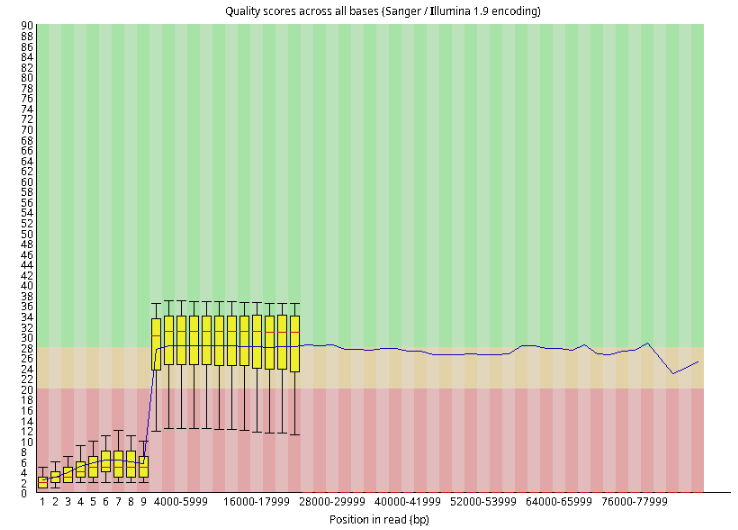
We could see the ONT read is way longer but contains more errors than the Illumina one.
Genome assembly with Illumina reads
Adapter removal with trimmomatic
trimmomatic is a tool for trimming adapters and low quality reads. [Read more]
The illumina reads are collected from the NextSeq platform, using the Nextera library preparation kit.
mkdir illumina
trimmomatic PE -threads 4 -phred33 data/wgs/NXT20x_R1.fastq.gz data/wgs/NXT20x_R2.fastq.gz illumina/NXT20x_R1_paired.fastq.gz illumina/NXT20x_R1_unpaired.fastq.gz illumina/NXT20x_R2_paired.fastq.gz illumina/NXT20x_R2_unpaired.fastq.gz ILLUMINACLIP:data/wgs/NexteraPE-PE.fa:2:30:10 LEADING:3 TRAILING:3 MINLEN:50
Expected output:
illumina/NXT20x_R1_paired.fastq.gz
illumina/NXT20x_R1_unpaired.fastq.gz
illumina/NXT20x_R2_paired.fastq.gz
illumina/NXT20x_R2_unpaired.fastq.gz
Reads quality control with bbmap
bbmap is a set of tools for quality control of sequencing reads. It can be used to remove the duplicated reads and reads from the PhiX control. [Read more]
clumpify.sh is a tool for removing duplicated reads. [Read more]
bbduk.sh is a tool for removing reads from contamination (E.g., host genome, the PhiX control). [Read more]
#clumpify
clumpify.sh in=illumina/NXT20x_R1_paired.fastq.gz in2=illumina/NXT20x_R2_paired.fastq.gz out=illumina/NXT20x_R1_paired_dedup.fastq.gz out2=illumina/NXT20x_R2_paired_dedup.fastq.gz dedupe optical spany adjacent
# bbduk
bbduk.sh in=illumina/NXT20x_R1_paired_dedup.fastq.gz in2=illumina/NXT20x_R2_paired_dedup.fastq.gz out=illumina/NXT20x_R1_paired_dedup_deduk.fastq.gz out2=illumina/NXT20x_R2_paired_dedup_deduk.fastq.gz ref=data/wgs/phiX174.fasta k=31 hdist=1
Expected output:
illumina/NXT20x_R1_paired_dedup.fastq.gz
illumina/NXT20x_R1_paired_dedup_deduk.fastq.gz
illumina/NXT20x_R2_paired_dedup.fastq.gz
illumina/NXT20x_R2_paired_dedup_deduk.fastq.gz
Genome assembly with spades
spades is a genome assembler for short reads. [Read more]
spades.py --isolate -t 4 -1 illumina/NXT20x_R1_paired_dedup_deduk.fastq.gz -2 illumina/NXT20x_R2_paired_dedup_deduk.fastq.gz -o illumina/spades
Expected assembly:
illumina/spades/contigs.fasta
Q2: We here assembled a bacterial genome from a isolate. What if we have a metagenomic sample?
We can use
--metaoption inspadesto assemble a metagenomic sample. [Read more]
Genome assembly with ONT reads
Optional: Adapter removal with guppy or porechop
Intallation of
guppyandporechopare not provided in this exercise. Try to install them by yourself if you want to use them.
guppy is a tool for basecalling and adapter trimming of ONT reads. guppy is not open-source, thus you need to register an ONT account for documentation and download. [Read more]
porechop is a open source tool for adapter trimming of ONT reads. [Read more]
By default, barcodes will be trimmed by guppy during the demultiplexing step. We will not repeat the barcode trimming on our data.
In case you want to trim the barcodes, you can use the following command for guppy.
guppy_barcoder -i data/wgs/ont_r10_20x.fastq.gz -s ont_r10/ont_r10_20x_barcoded.fastq.gz --barcode_kits EXP-NBD104 --trim_barcodes
And the following command for porechop.
porechop -i data/wgs/ont_r10_20x.fastq.gz -o ont_r10/ont_r10_20x_porechop.fastq.gz --threads 4
Reads quality control with seqkit
seqkit is a tool for manipulating sequencing data. [Read more] Here we use seqkit to remove the short reads and reads with low quality.
mkdir ont_r10
seqkit seq -j 4 -Q 10 -m 2000 -i data/wgs/ont_r10_20x.fastq.gz -o ont_r10/ont_r10_20x_f.fastq.gz
Check ONT reads before and after quality control with seqkit stat.
seqkit stat data/wgs/ont_r10_20x.fastq.gz ont_r10/ont_r10_20x_f.fastq.gz
Expected output:
file format type num_seqs sum_len min_len avg_len max_len
data/wgs/ont_r10_20x.fastq.gz FASTQ DNA 7,862 51,201,670 129 6,512.6 87,688
ont_r10/ont_r10_20x_f.fastq.gz FASTQ DNA 6,561 48,742,515 2,001 7,429.1 87,688
Genome assembly with flye
flye is a long-read genome assembler, recommended by ONT. [Read more]
flye --nano-raw ont_r10/ont_r10_20x_f.fastq.gz --out-dir ont_r10/flye --threads 4
Expected assembly file:
ont_r10/flye/assembly.fasta
Q3: By default,
flyeis used to assembly genomes. What if we have a metagenomic sample?We can use
--metaoption inflyeto assembly a metagenomic sample. [Read more]
Genome polishing with racon and medaka
long-read genome assemblers usually produce a draft genome with high contiguity but low accuracy. Extra polishing steps are needed to improve the accuracy of the draft genome. But the best polishing stratefy and tools are still under debate.
racon is a graph-based consensus algorithm for polishing long-read genome assemblies. [Read more].
medaka is a official ONT polishing tool based on neural network. [Read more]
Recently, the ONT recommended directly polishing flye assembly with medaka. But combining racon and medaka is still a common practice.
Here we only choose medaka for polishing in our example.
Since medaka is based on neural network, choosing the appropriate model will affect the polishing result. You can use medaka tools list_models to list all the available models. For our example, the ONT reads are collected from the R10.4.1 flowcell, and the hac mode of guppy is used for basecalling.
medaka tools list_models
medaka_consensus -i ont_r10/ont_r10_20x_f.fastq.gz -d ont_r10/flye/assembly.fasta -o ont_r10/medaka -t 4 -m r1041_e82_260bps_hac_v4.1.0
Expected assembly file:
ont_r10/medaka/consensus.fasta
Optional: polishing with racon and medaka
mkdir ont_r10/racon
minimap2 -t 4 -x map-ont ont_r10/flye/assembly.fasta ont_r10/ont_r10_20x_f.fastq.gz > ont_r10/racon/flye_assembly.paf
racon -t 4 ont_r10/ont_r10_20x_f.fastq.gz ont_r10/racon/flye_assembly.paf ont_r10/flye/assembly.fasta > ont_r10/racon/racon.fasta
medaka_consensus -i ont_r10/ont_r10_20x_f.fastq.gz -d ont_r10/racon/racon.fasta -o ont_r10/racon/medaka -t 4 -m r1041_e82_260bps_hac_v4.1.0
Expected assembly file:
ont_r10/racon/racon.fasta
ont_r10/racon/medaka/consensus.fasta
Hybrid assembly with ONT and Illumina reads
Hybrid assembly is a common practice to combine the advantages of different sequencing technologies. Here we choose two commonly adopted strategies for hybrid assembly:
- Directly use the ONT assembly as the backbone and polish it with Illumina reads.
pilon - Short-read-first hybrid assembly.
unicycler
Illumina reads polishing with pilon
pilon is a tool for polishing genome assemblies with short reads. [Read more]
mkdir -p hybrid/pilon
bwa index ont_r10/medaka/consensus.fasta
bwa mem -t 4 ont_r10/medaka/consensus.fasta illumina/NXT20x_R1_paired_dedup_deduk.fastq.gz illumina/NXT20x_R2_paired_dedup_deduk.fastq.gz | samtools sort -@ 4 -o hybrid/pilon/aligned.bam
samtools index hybrid/pilon/aligned.bam
pilon --genome ont_r10/medaka/consensus.fasta --frags hybrid/pilon/aligned.bam --output hybrid/pilon/pilon --threads 4
Expected assembly file:
hybrid/pilon/pilon.fasta
Optional: hybrid assembly with unicycler
unicycler can conduct a short-read-first hybrid assembly. [Read more]
It takes some time, so we will skip this step in the example. Feel free to come back to this step after you have finished the other steps.
Linux users
unicycler -l ont_r10/ont_r10_20x_f.fastq.gz -1 illumina/NXT20x_R1_paired_dedup_deduk.fastq.gz -2 illumina/NXT20x_R2_paired_dedup_deduk.fastq.gz -o hybrid/unicycler --threads 4
MacOs users
Since the latest MacOS version of unicycler on conda is behind 0.5.0, we append two flags of --no_correct and --no_pilon to maintain maximum compatibility.
unicycler --no_correct --no_pilon -l ont_r10/ont_r10_20x_f.fastq.gz -1 illumina/NXT20x_R1_paired_dedup_deduk.fastq.gz -2 illumina/NXT20x_R2_paired_dedup_deduk.fastq.gz -o hybrid/unicycler
Expected assembly file:
hybrid/unicycler/assembly.fasta
Quality assessment of assembled genomes with quast
We have generated many assemblies with different strategies. Let’s use a deeply sequenced PacBio assembly as the reference genome to assess the quality of the assemblies. You can take the provided assemblies under ./data/wgs/assemblies if you haven’t finished all assembly steps.
seqkit stat data/wgs/assemblies/*.fasta data/wgs/ncbi_pacbio_TL110.fasta
Expected output:
file format type num_seqs sum_len min_len avg_len max_len
data/wgs/assemblies/hybrid_pilon.fasta FASTA DNA 2 2,579,926 29,242 1,289,963 2,550,684
data/wgs/assemblies/hybrid_pilon_proovframe.fasta FASTA DNA 2 2,579,979 29,245 1,289,989.5 2,550,734
data/wgs/assemblies/hybrid_unicycler.fasta FASTA DNA 1 2,564,177 2,564,177 2,564,177 2,564,177
data/wgs/assemblies/illumina.fasta FASTA DNA 217 2,527,918 78 11,649.4 60,978
data/wgs/assemblies/ont_flye.fasta FASTA DNA 2 2,579,333 29,239 1,289,666.5 2,550,094
data/wgs/assemblies/ont_flye_medaka.fasta FASTA DNA 2 2,579,052 29,238 1,289,526 2,549,814
data/wgs/assemblies/ont_flye_racon.fasta FASTA DNA 2 2,578,932 29,231 1,289,466 2,549,701
data/wgs/assemblies/ont_flye_racon_medaka.fasta FASTA DNA 2 2,578,566 29,229 1,289,283 2,549,337
data/wgs/ncbi_pacbio_TL110.fasta FASTA DNA 1 2,566,312 2,566,312 2,566,312 2,566,312
Compared to the reference, the total length of the assemblies are similar.
But the number of sequences (
num_seqs) and the maximal sequence length (max_len) vary a lot.
Let’s chcek the quality of the assemblies with quast.
quast data/wgs/assemblies/*.fasta -r data/wgs/ncbi_pacbio_TL110.fasta -o quast
You can open the report.html file in the quast directory to have a look at the quality of the assemblies. 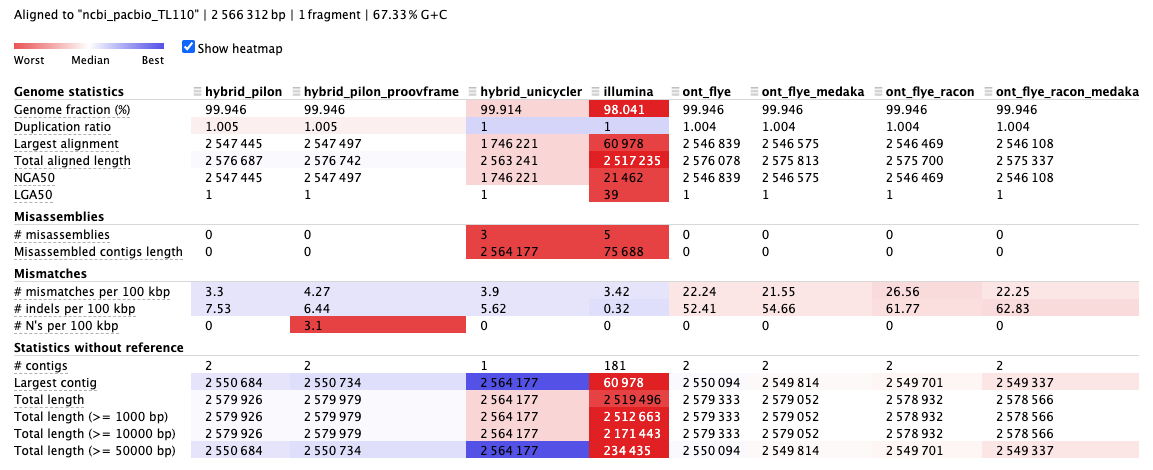
Q4: Based on the example, which sequencing technology would you think works better in genome completeness and contiguity?
Check
Genome fractionandNGA50Q5: Based on the example, which assembly do you think is the best in accuracy?
Check
MisassembliesandMismatchesQ6: Based on the example, what is the dominant error type in the ONT assembly?
Check
mismatchesandindels. [Read more]Q7: What would you think is the best assembly strategy for this example?
Optional: Reference-guided correction with proovframe
High frequency of indels results in frameshifts in the coding sequences (CDSs) of the ONT assembly. With reference-guided correction, we can correct the frameshifts in the CDSs of the ONT assembly.
This step is optional, you can skip it if you don’t have time.
To use the proovframe tool, we need to create another conda environment with mamba due to the dependency conflict.
conda deactivate
mamba env create -n wgs2 -f envs/env2.yaml
conda activate wgs2
Genome annotation with prokka
prokka is a commonly used tool for rapid annotation of bacterial genomes. [Read more]
mkdir proovframe
conda activate wgs2
prokka --outdir proovframe/prokka --prefix pacbio --cpus 4 data/wgs/ncbi_pacbio_TL110.fasta
Expected output:
proovframe/prokka/pacbio.err
proovframe/prokka/pacbio.faa
proovframe/prokka/pacbio.ffn
proovframe/prokka/pacbio.fna
proovframe/prokka/pacbio.fsa
proovframe/prokka/pacbio.gbk
proovframe/prokka/pacbio.gff
proovframe/prokka/pacbio.log
proovframe/prokka/pacbio.sqn
proovframe/prokka/pacbio.tbl
proovframe/prokka/pacbio.tsv
proovframe/prokka/pacbio.txt
We have many output files from prokka. Here we only use the translated protein sequences (./proovframe/prokka/pacbio.faa file) for proovframe.
Frameshift correction with proovframe
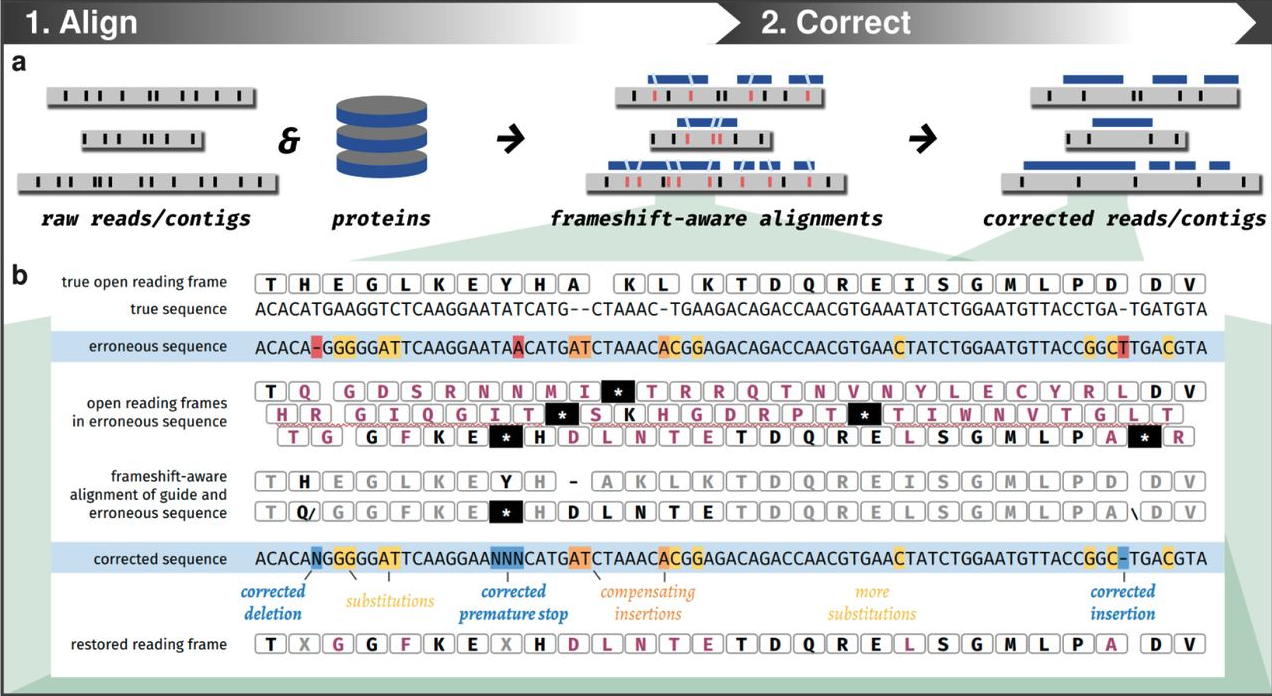
proovframe is a tool for reference-guided correction of frameshifts in CDSs. [Read more]
proovframe map -a proovframe/prokka/pacbio.faa -o proovframe/pilon.tsv hybrid/pilon/pilon.fasta
proovframe fix -o proovframe/pilon_corrected.fasta hybrid/pilon/pilon.fasta proovframe/pilon.tsv
Expected assembly file:
proovframe/pilon_corrected.fasta
Q8: In the previous
quastreport, there’s an increase ofN'sin thehybrid_pilon_proovframe.fastaassembly. Do you think it’s good or bad?